肿瘤免疫治疗的耐药机制(整理:王欢)
整理自公众号:科睿唯安生命科学与制药
1 肿瘤免疫治疗药物现状
2014年anti-PD-1单抗Opdivo和Keytruda批准上市以来,肿瘤免疫治疗的热浪席卷全球,取得了空前的成功。除Opdivo和Keytruda外,还有anti-CTLA-4单抗Yervoy以及anti-PD-L1单抗Tecentriq获FDA批准上市。这些免疫检查点抑制剂在黑色素瘤,非小细胞肺癌,肾细胞癌,霍奇金淋巴瘤,膀胱癌等多种癌症中表现出了令人欣喜的疗效,其中Keytruda还在2016年获批一线治疗PD-L1高表达(>50%)的非小细胞肺癌。
PD-1/PD-L1抗体药物是肿瘤免疫疗法的里程碑,市场表现也是一再超出预期,2016年PD-1/PD-L1市场规模已达60亿美元。
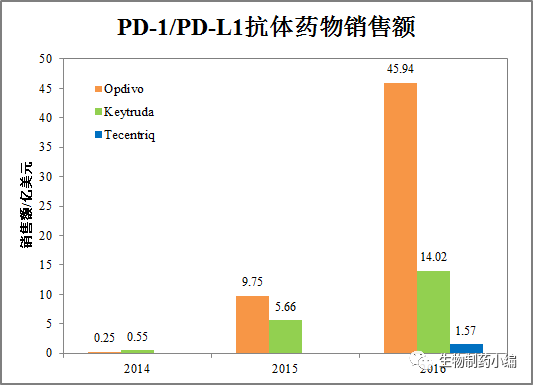
图1 PD-1/PD-L1抗体药物销售额
2 肿瘤免疫治疗的耐药机制
免疫检查点抑制剂并不是万能的“神药”,也存在其缺陷,其中一项就是由于耐药性而导致的响应率不高与肿瘤复发再进展。肿瘤免疫耐药可分为原发性耐药(primary resistance),适应性免疫耐药(adaptive immune resistance)以及获得性耐药(acquired resistance)。在anti-PD-1疗法治疗黑色素瘤中,约有60%的患者不响应(primary resistance),还有一部分患者在初始响应后肿瘤出现了再进展。
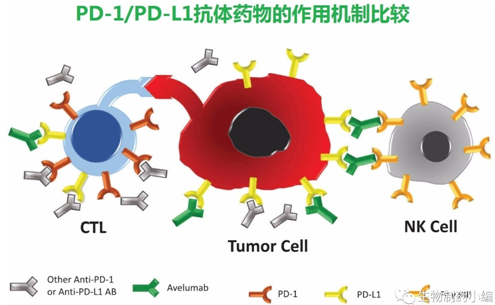
图2 PD-1/PD-L1抗体药物的作用机制比较
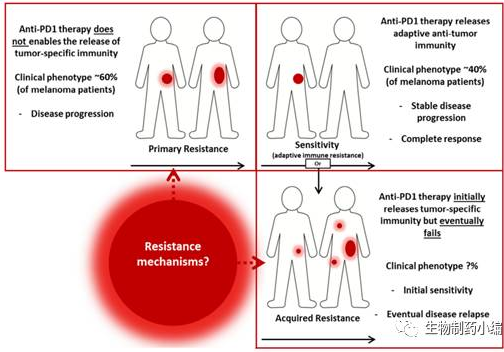
图3 黑色素瘤患者对anti-PD-1疗法的临床响应
2.1 导致原发性/继发性耐药的肿瘤内在原因
导致原发性/继发性耐药的肿瘤内在原因主要是肿瘤细胞上特定基因或通路的表达或上调,从而导致肿瘤微环境中免疫细胞的浸润以及功能受到抑制。具体如下:
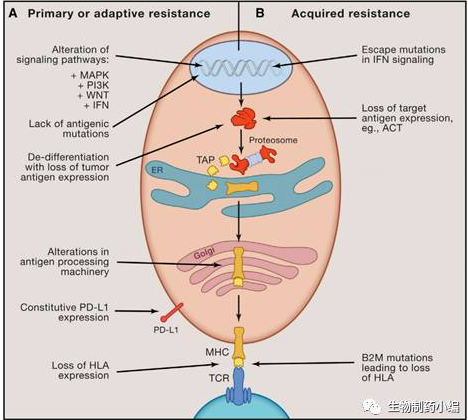
图4 导致免疫疗法耐药的肿瘤内在原因
2.1.1 MAPK通路的激活与或PTEN表达的缺失而引起的PI3K通路的增强
癌基因信号通过MAPK通路导致VEGF与IL-8的产生,从而抑制T细胞的招募与功能。此外,在多种肿瘤中,肿瘤抑制基因PTEN表达缺失从而PI3K通路增强,这与IFNγ,颗粒酶B的基因表达量降低以及肿瘤浸润CD8+ T细胞的数目减少是高度相关的。
2.1.2 WNT/β-catenin信号通路的持续表达
癌基因信号通过稳定β-catenin导致WNT信号通路持续激活,从而将T细胞排除在肿瘤之外。在人Non-T-cell-inflamed的黑色素瘤中,肿瘤内在的β-catenin信号基因高度表达,且在肿瘤微环境中缺少T细胞与CD103+ DC细胞。
2.1.3 肿瘤上PD-L1的高表达
肿瘤上高表达的某些配体如PD-L1,会抑制抗肿瘤T细胞的应答。多种机制可能导致PD-L1高表达,包括PTEN的缺失或PI3K/AKT的突变,EGFR突变,MYC过表达,CDK5基因破坏以及PD-L1基因3’-UTR的截短。目前还不知道PD-L1的高表达是否会对anti-PD-1/PD-L1的响应有影响,但是它会影响其他的肿瘤免疫疗法。
2.1.4 IFNγ信号通路的缺失
由肿瘤特异的T细胞产生的IFNγ,能够识别肿瘤细胞或抗原递呈细胞上的相应受体,从而发挥有效的抗肿瘤免疫响应。IFNγ能够增强MHC分子的表达,从而增强肿瘤抗原提呈作用。IFNγ也能够招募其他的免疫细胞,或者直接抑制肿瘤细胞的增殖,促进其凋亡。因此肿瘤细胞上IFNγ通路相关蛋白,如IFNγ受体IFNGR1与IFNGR2,IFNγ受体链JAK1与JAK2,STATs,IRF1等突变与缺失,都会导致对免疫检查点抑制剂的耐药。
2.1.5 缺少肿瘤抗原,从而导致T细胞无法激活
免疫疗法依赖于肿瘤抗原特异的T细胞。在人黑色素瘤,肾细胞癌,非小细胞肺癌中,DNA突变频率高,肿瘤免疫原性更强,因而对anti-PD-1疗法响应更好。而在胰腺癌以及前列腺癌中,DNA突变频率低,肿瘤免疫原性低,对anti-PD-1疗法响应差。
2.1.6 抗原提呈机制存在缺陷
在某些情况下,由于抗原加工过程中的蛋白酶体成员,转运蛋白,MHC本身以及beta-2-微球蛋白(B2M)的功能缺陷,会导致抗原提呈机制不能有效地将肿瘤抗原提呈到细胞表面。B2M在HLAI家族的折叠与转运到细胞膜的过程中发挥关键作用,若其丧失功能,则CD8+T细胞失去了识别功能。
2.1.7 存在一系列特定基因的表达
在对PD-1疗法没有响应的肿瘤中,有一些基因表达被富集,被称为innate anti-PD-1 resistance signature,或IPRES。这些基因与间叶细胞的转化,全能型以及伤口愈合相关,且更倾向于表达在胰腺癌等对PD-1不响应的肿瘤中。
2.2 导致原发性/继发性耐药的肿瘤外部原因
导致原发性/继发性耐药的肿瘤外部原因是由于肿瘤微环境中一些成员发挥抗癌免疫响应的抑制作用,主要包括调节T细胞Tregs,髓样抑制细胞MDSCs,M2巨噬细胞,其他的抑制性免疫检查点与抑制性细胞因子等。
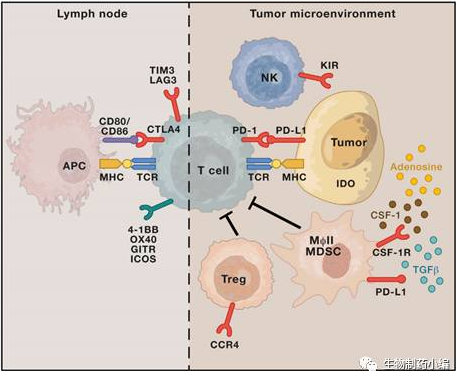
图5 导致免疫疗法耐药的肿瘤外部原因
2.2.1 Tregs
Tregs能通过分泌抑制性细胞因子或者通过直接的细胞接触来抑制效应T细胞Teffs的响应。许多人肿瘤中发现了浸润的Tregs,且鼠模型中去除肿瘤微环境中的Tregs能够显著提高免疫响应。由于CTLA-4在Tregs上高表达,anti-CTLA-4能够显著提高Teffs/Tregs的比例,从而提高肿瘤对免疫疗法的响应。
2.2.2 Myeloid-derived suppressor cells (MDSCs)
MDSCs在多种病理条件包括肿瘤,发挥着免疫响应调节因子的作用。人MDSCs表达CD11b+与CD33+,但是不表达HLA-DR以及系种特异的抗原CD3,CD19与CD57。MDSCs能够促进血管生长,肿瘤侵袭与转移。肿瘤微环境中MDSCs的存在与降低的生存率以及免疫检查点抑制剂疗法的响应率相关。目前实验中使用PI3Kγ抑制剂来调节巨噬细胞功能,在老鼠模型中,PI3Kγ抑制剂与anti-PD-1联用表现出了良好的肿瘤抑制效果。
2.2.3 M2 macrophages
肿瘤相关的巨噬细胞(Tumor-Associated Macrophages,TAMs)也能够影响免疫治疗的响应。TAMs包括M1巨噬细胞和M2巨噬细胞,在大多数情况下M2巨噬细胞占TAMs的大多数。其中M1巨噬细胞能够高表达IL-12,IL-23,MHC以及B7家族分子来促进抗原提呈与Th1细胞的激活,从而发挥抗肿瘤免疫作用;而M2巨噬细胞能够分泌抑制性细胞因子IL-10与TGF-β,从而抑制免疫响应与促进肿瘤生长与转移。临床上TAMs的数目越多,肿瘤预后就越差。临床前实验使用巨噬细胞集落刺激生长因子受体1(CSF-1R)的抑制剂,能够显著减少TAMs数目,抑制肿瘤生长。CSF-1R抑制剂与anti-CTLA4或anti-PD1再加上吉西他滨联用,能够有效缓解单独anti-CTLA-4或anti-PD1不响应的鼠胰腺癌模型。
2.2.4 其他的抑制性免疫检查点
除了PD-1与CLTA-4,T细胞上还存在其他的抑制性免疫检查点,包括TIM-3,LAG-3,BTLA,TIGIT,和VISTA等。
2.2.5 免疫抑制细胞因子与免疫抑制分子
肿瘤或者巨噬细胞会释放一些免疫抑制细胞因子或者免疫抑制分子来减弱局部的抗肿瘤免疫反应。TGF-β能够促进血管生成,刺激Tregs从而发挥免疫抑制作用。在多种肿瘤中,高水平的TGF-β都伴随着极差的预后。临床前实验使用TGF-β受体激酶抑制剂与anti-CTLA-4联用,或者放疗与TGF-β抑制剂都显示出了较好的肿瘤抑制效果。在细胞外,CD39能够将ATP水解成AMP,进一步被胞外核苷酶CD73加工为免疫抑制分子腺苷adenosine。Adenosine能够通过T细胞上的A2A受体抑制T细胞的增殖与细胞毒活性,也能通过肿瘤细胞上的A2B受体促进肿瘤转移。多种类型的肿瘤中,CD73的高表达伴随着较差的预后,且会影响anti-PD-1的效果。此外,IFNγ会促进免疫抑制分子IDO的表达,IDO能直接负调控效应T细胞的功能。
2.2.6 趋化因子与趋化因子受体
某些特异的趋化因子与趋化因子受体在MDSCs和Tregs往肿瘤微环境的运输过程中起到重要作用。肿瘤细胞能够分泌配体CCL5,CCL7和CXCL8,通过结合MDSCs上表达的CCR1及CXCR2受体,从而将MDSCs吸引至肿瘤微环境中。CCR4在Tregs上高表达,anti-CCR4能够有效抑制T细胞的招募并通过ADCC效应减少Tregs的数目。此外CXCR4是CXCL12的受体,CXCL12能够通过影响Tregs定位等多种方式发挥免疫抑制作用。
2.2.7 肿瘤浸润细胞上CD28的表达
CD28是T细胞的共刺激分子,对于T细胞的激活,增殖和存活起到关键作用。在人出生时,所有T细胞都会表达CD28,但是CD28的表达量会随着年龄的增加而下降,80岁时会有10-15%的CD4+ T细胞以及50-60%的CD8+ T细胞缺失CD28表达。CD28-T细胞产生的主要原因是重复的抗原刺激。CD28表达的消失只存在与人和灵长动物中,在鼠中并没有被发现。在今年3月9日的《Science》上,发表了来自Emory疫苗中心的研究工作,他们发现CD28对于耗尽的CD8+T细胞(Exhausted CD8+ T Cell)的再激活是必须的。在老鼠模型中,阻断CD28-B7共刺激通路,会影响肿瘤特异CD8+T细胞的增殖和激活,降低对anti-PD-1/PD-L1疗法的响应。在经过anti-PD-1治疗的NSCLC病人中,增殖的CD8+T细胞(高Ki-67表达)大都为PD-1阳性,且被激活(高HLA-DR,CD38表达)。和鼠模型中一样,这些增殖的CD8+T细胞大都是CD28阳性的,说明了CD28共刺激对于肿瘤浸润的PD-1+CD8+T细胞的增殖与再激活是至关重要的。因此,CD28可以用作预测anti-PD-1疗法响应程度的分子标记。
2.3 获得性耐药的产生原因
肿瘤免疫疗法的一个显著特点是肿瘤响应的时间长,但是在使用anti-CTLA-4或anti-PD-1治疗黑色素瘤的患者中,大约有1/4-1/3的患者在经历初始响应后出现了肿瘤进展,即获得性耐药。获得性耐药的产生可能是因为在治疗过程中出现了上文所述的那些原因,包括T细胞功能的丧失,肿瘤抗原提呈出现缺陷以及其他一些新突变的产生。例如,在一类较晚获得anti-PD-1耐药性的病人中,发现了B2M的突变,从而导致抗原提呈机制缺陷。在另两例获得性耐药肿瘤中,发现了IFNγ通路中JAK1或JAK2发生了突变。
在2016年《nature communication》上,有研究者发现anti-PD-1耐药性的产生与抑制性免疫检查点TIM-3的的表达量升高密切相关。在两个老鼠肿瘤模型中,耐药后与给药前相比,肿瘤浸润的T细胞而非外周血或脾脏T细胞上的TIM-3表达量显著上调,且TIM-3表达量上调的主要是那些结合了anti-PD-1抗体的T细胞。需注意的是TIM-3表达量升高是anti-PD-1疗法特异的,因为anti-CTLA-4中并未检测到类似现象。此外,肿瘤细胞上TIM-3配体Galectin-9的表达量也显著升高。当anti-PD-1疗法出现耐药后,联用anti-TIM-3,显著提高了生存率。有意思的是,当anti-PD-1和anti-TIM-3联用被耐药,肿瘤重新进展时,那些结合了anti-PD-1和anti-TIM-3抗体的T细胞上,其他的抑制性免疫检查点如CTLA-4,LAG-3的表达量明显升高。这说明了肿瘤浸润T细胞的抑制性免疫调节是动态变化的,存在着补偿效应。最后,在两例对anti-PD-1疗法获得性耐药的NSCLC病人肿瘤样本中,也观察到了TIM-3而非其他抑制性免疫检查点的表达量显著升高。这些结果说明了anti-PD-1和anti-TIM-3联用是对于那些anti-PD-1疗法获得性耐药病人的一个良好策略。
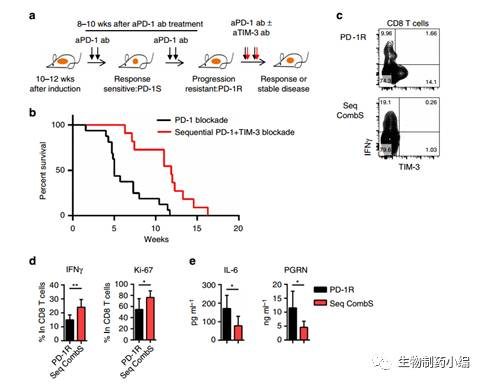
图6 Anti-TIM-3与anti-PD-1联用能显著抑制肿瘤进展
3 应对肿瘤免疫耐药的方案
随着精准医疗的概念深入人心,鉴定出有效的生物标记来预测免疫疗法的响应与耐药非常有意义。这些生物标记包括基因组标记,免疫调节基因表达量以及浸润CD8+T细胞的密度和分布等免疫标记。相对于之前使用的治疗前本底水平的生物活检(pre-treatment baseline biopsies),纵向的多次活检(longitudinal biopsies)可能会更好地预测肿瘤对治疗的响应。最近研究表明相比于治疗前的标记,治疗早期样本的免疫标记有更好地预测效果。
此外,应对肿瘤免疫耐药的一个有效方法就是联合用药。Anti-PD-1与anti-CTLA-4联合用药治疗转移性黑色素瘤显示出了更高的响应率。Anti-PD1与anti-TIM-3/anti-LAG-3/anti-TIGIT的联合用药在临床前实验都展示出了极佳的效果,临床试验也正在进行中。除了免疫检查点抑制剂之间联用,免疫检查点抑制剂与共刺激因子激动剂,靶向药,放疗化疗以及表观遗传修饰因子的联用都在探索之中。希望未来免疫检查点抑制剂能够联合一些便宜的非专利药,来减少联合用药带来的高花费。
